Chapter 5 Heterozigosity analysis
heterozigosity <- read_tsv("data/heterozigosity.tsv") %>%
rename(mouse=1,obs_hom=2,est_hom=3,n_sites=4,f_stat=5) %>%
mutate(obs_het=n_sites - obs_hom) %>%
mutate(est_het=n_sites - est_hom) %>%
mutate(obs_het_perc=obs_het / n_sites * 100) %>%
mutate(est_het_perc=est_het / n_sites * 100) %>%
mutate(cage=substr(mouse, 1, 3))
shapiro.test(heterozigosity$obs_het_perc)
heterozigosity %>%
as.data.frame() %>%
aov(obs_het_perc ~ cage, data = .) %>%
broom::tidy()
heterozigosity %>%
ggplot(aes(y=cage,x=obs_het_perc,group=cage))+
geom_boxplot(fill="#f4f4f4")+
theme_minimal()
mean_heterozigosity <- heterozigosity %>%
group_by(cage) %>%
summarise(mean=mean(obs_het_perc),sd=sd(obs_het_perc))
heterozigosity %>%
group_by(cage) %>%
summarise(mean=mean(obs_het_perc),sd=sd(obs_het_perc)) %>%
tt()heterozigosity %>%
ggplot(aes(y=fct_rev(cage),x=obs_het_perc,group=cage))+
geom_boxplot(fill="#f4f4f4", color="#666666")+
theme_minimal()
hierarchy_stability %>%
as.data.frame() %>% t() %>% t() %>% as.data.frame() %>%
rownames_to_column(var="cage") %>%
rename(cage=1,stability=2) %>%
left_join(mean_heterozigosity, by=join_by(cage==cage)) %>%
{cor.test(.$stability, .$mean)}hierarchy_stability %>%
as.data.frame() %>% t() %>% t() %>% as.data.frame() %>%
rownames_to_column(var="cage") %>%
rename(cage=1,stability=2) %>%
left_join(mean_heterozigosity, by=join_by(cage==cage)) %>%
ggplot(aes(x=mean,y=stability)) +
geom_point() # GWAS analysis
# GWAS analysis
5.1 Association analysis
This step is repeated for each time point separately.
# SNP data
X = read.plink("data/variants_filtered.bed")
X = as(X$genotypes,'numeric')
# Metadata
data = read_table("data/dominance_t1.tsv") %>%
mutate(cage=factor(cage)) %>%
mutate(mouse=factor(mouse)) %>%
as.data.frame()
# Run GWAS
gwas = GridLMM_GWAS(formula = dominance ~ 1 + (1|cage),
test_formula = ~1,
reduced_formula = ~0,
data = data,
X = X,
X_ID = 'mouse',
method = 'REML',
fillNAX = TRUE,
verbose = F,
mc.cores = 8)5.1.1 Manhattan plot
This step is repeated for each gwas result separately.
# Prepare working table
gwas_result <- gwas$results %>%
#slice_sample(n=100000) %>%
separate(X_ID, into = c("chr", "bp"), sep = "_", remove = FALSE) %>% # separate chromosome and position
mutate(bp=as.numeric(bp)) %>%
mutate(chr=factor(chr, levels=c(c(1:19),"X"))) %>%
rename(p=p_value_REML) %>%
filter(!is.nan(p))
###
# Custom Manhattan
###
# https://r-graph-gallery.com/101_Manhattan_plot.html
# Append chromosome coordinates
gwas_result <- gwas_result %>%
group_by(chr) %>%
summarise(chr_len=max(bp)) %>%
mutate(tot=cumsum(chr_len)-chr_len) %>%
select(-chr_len) %>%
left_join(gwas_result, ., by=c("chr"="chr")) %>%
arrange(chr, bp) %>%
mutate(bp_cum=bp+tot)
# Append significance level
gwas_result_unsig <- gwas_result %>%
filter(p > 0.000001)
# Generate chromosome number code
axisdf <- gwas_result %>%
group_by(chr) %>%
summarize(center=mean(bp_cum))
# Plot gwas
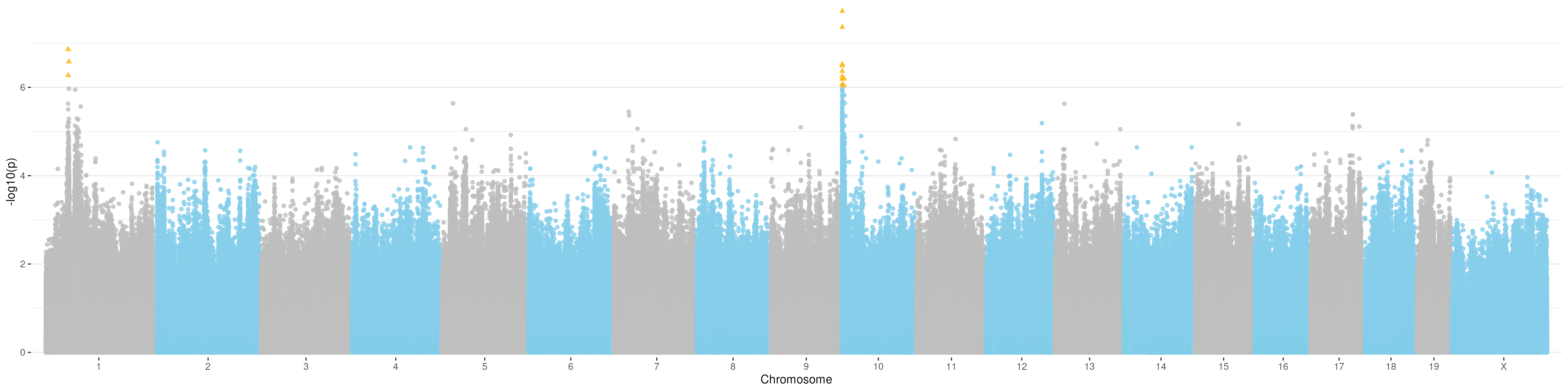
ggplot(gwas_result_unsig, aes(x=bp_cum, y=-log10(p))) +
# Show all points
geom_point(aes(color=as.factor(chr)), alpha=0.8, size=1.3) +
scale_color_manual(values = rep(c("grey", "skyblue"), 22 )) +
# Show significant points
geom_point(data=gwas_result_sig, aes(x=bp_cum, y=-log10(p)), color="#FFB91F", shape=17, alpha=0.9, size=1.6) +
# custom X axis:
scale_x_continuous(label = axisdf$chr, breaks= axisdf$center, expand = c(0.01, 0.01)) +
scale_y_continuous(expand = c(0.01, 0.04)) +
# Custom the theme:
theme_bw() +
labs(x="Chromosome") +
theme(
legend.position="none",
panel.border = element_blank(),
panel.grid.major.x = element_blank(),
panel.grid.minor.x = element_blank()
)
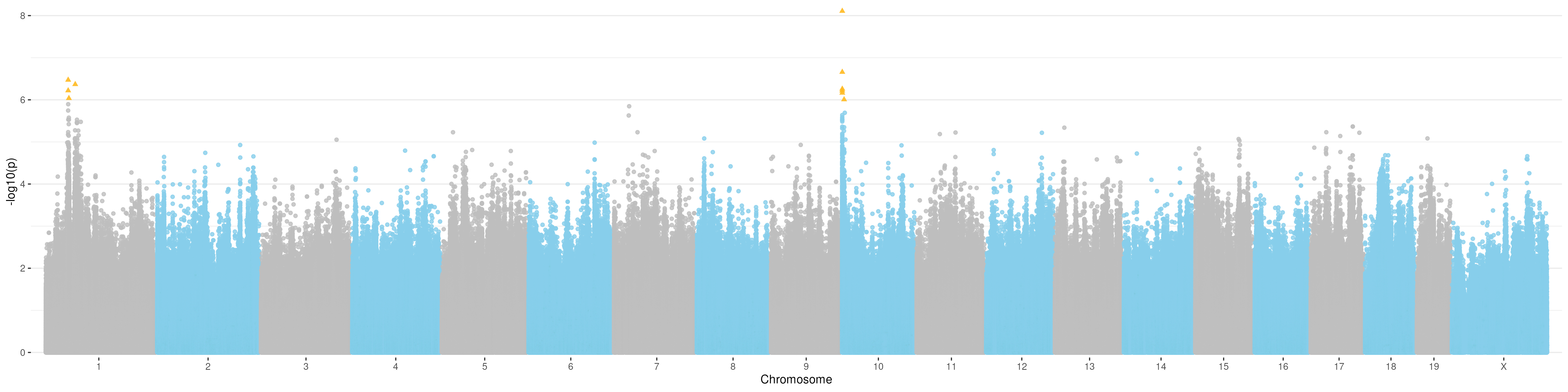
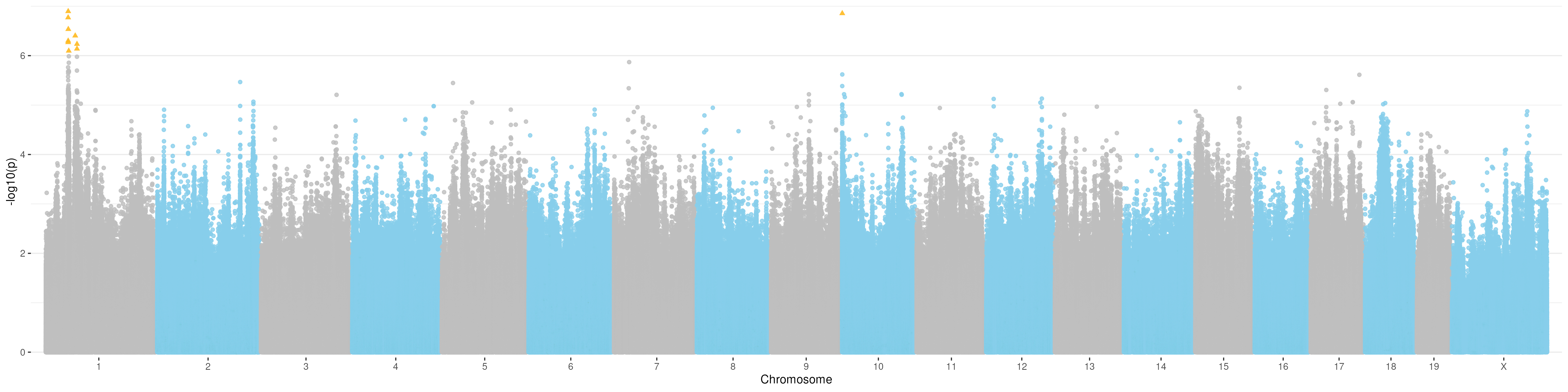
Manhattan plot for t3
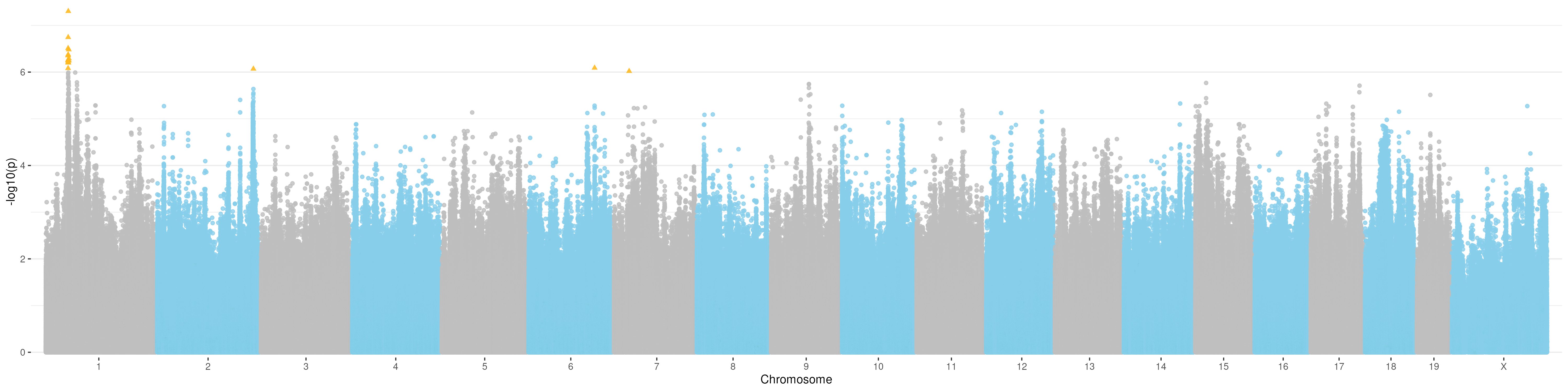
Manhattan plot for t4
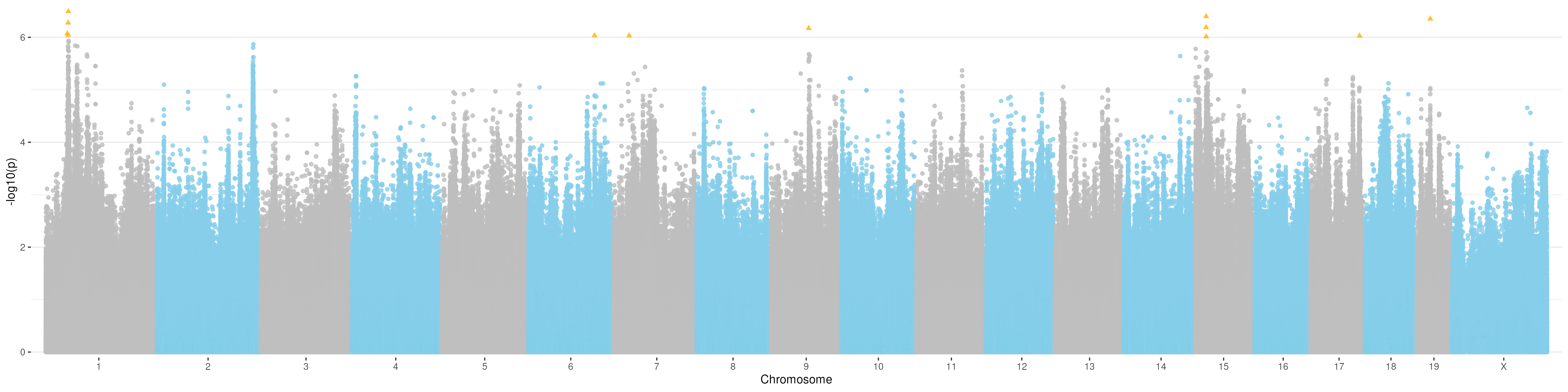
Manhattan plot for t5
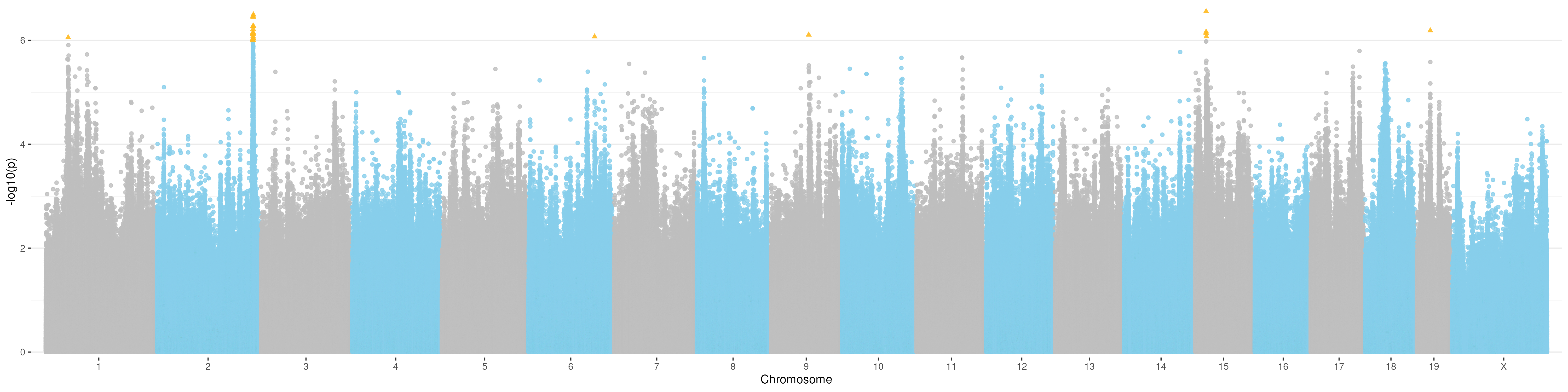
Manhattan plot for t6